DNAGenre
AutumnMiles: DNAGenre
This project was aimed at being free, used by organizations, and a startup project for study purposes. It is aimed at making DNA analysis faster and more insightful (visionary).
Dive Into
About
DNAGenre is a bioinformatics (study of DNA using computers) oriented software that helps to read a DNA/RNA file and provide insight in DNA/RNA data. This software is different in away that for the start, it is educational, free to use, and will commonly be used in labs. The software logs in a user or signs up a user, then they can just tap a real DNA/RNA file, and the software will provide the insights of the DNA file. The problem the project solves is that it is educational and helps learners know what is bioinformatics under the hood.
Note: This project was 35% AI and 65% me
Features
- Login and User Authentication
- Reading Folder for sequence files
- Using real bioinformatic files (good with
txtfiles in required format) - Bioinformatic analysis
- Codon Analysis
- Protein Analysis
- Simple Inbuilt Programming Language called
Decode - User can delete a created account.
- Show time on some processes.
- Able to process an entire folder of DNA/RNA files.
Note: Run only the GUI.py file to see the project in action.
Bugs Found
DataTools.py
def clean_data(filepath, label_char):
"""
Receives a file path and separates strand labels from DNA
Returns a dict of {strand labels: DNA}
"""
DNAdict = dict()
# Read Index, If No Read Index Then '?'
label = '?'
with open(filepath, 'r') as file:
for i in file.readlines():
i = i.strip()
if label_char in str(i):
label = i
DNAdict[label] = ''
else:
DNAdict[label] = ''
DNAdict[label] += i
return DNAdict
Note: This function has a bug and as we go on, we will see the bug.
1. Imagine A File Like This
TTGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
GGGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@1
ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@2
GTGGCGTGAGTGCGGATCGATCGATGATCGATTCGATCGATCGCGCGAAAATAGAGCTAGCCC
@3
TCTTGATCGATCGATCGCTATTAGACGGCATGATCGATTCGATCGATCGCGCGAAAATATAGC
@4
ATCGTACCAGTACATACGATAGCCTCGTGATTCGATCGATGTGGCGTGAGTGCGGATCGATCG
@5
GTGGCAATATAGTGAGAGTAGAGTGCGGATCGAAATCGCTATCTAAGCTCAGTCGATTCTCTA
@6
CTAGACAGCTACAGTCCATGCGTGGCGTGAGCGATAGCTATCGATGACTCGATTGCGGATCGG
@7
GGTGAGTGCGGATCCGCTAGCTACCACATACGGGATAAAGCTAGTCTCTACGACGGTAGAGTA
@8
AGCTACGATTTATATCGGCTAGCCGTTATATAGCCGGACACAGATAGTACACAACAGTAGAGT
@9
GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC
@10
GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
When clean_data function is called, it makes label equal ?. Now in this scenario, it finds DNA first but not label
hence label is set to ? and DNA is collected. On the next run, the label is set to ? but when we enter this line
if label_char in str(i): there is no label_char (for this scenario label_char is @) so it jumps to else and
else sets label to '' (so our previous ?:'TTGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA' is
lost and now is '?':'' and the next line of code does add to the current dict key hence
'?':'GGGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA') and the code go on smoothly as intended.
Note: This function has a bug and as we go on, we **will see the bug.**
2. Imagine Another Like This
TTGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
GGGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@1ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@2GTGGCGTGAGTGCGGATCGATCGATGATCGATTCGATCGATCGCGCGAAAATAGAGCTAGCCC
@3TCTTGATCGATCGATCGCTATTAGACGGCATGATCGATTCGATCGATCGCGCGAAAATATAGC
@4ATCGTACCAGTACATACGATAGCCTCGTGATTCGATCGATGTGGCGTGAGTGCGGATCGATCG
@5GTGGCAATATAGTGAGAGTAGAGTGCGGATCGAAATCGCTATCTAAGCTCAGTCGATTCTCTA
@6CTAGACAGCTACAGTCCATGCGTGGCGTGAGCGATAGCTATCGATGACTCGATTGCGGATCGG
@7GGTGAGTGCGGATCCGCTAGCTACCACATACGGGATAAAGCTAGTCTCTACGACGGTAGAGTA
@8AGCTACGATTTATATCGGCTAGCCGTTATATAGCCGGACACAGATAGTACACAACAGTAGAGT
@9GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC
@10GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
When clean_data function is run, it will function as explained earlier but when reaches this part
@1ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA this code if label_char in str(i):runs, this
creates the paradox of having keys equal to @1ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGAand
values equal to '' hence a result of Read Index plus nucleotide chars:''respectively.
3. Imagine Another Third Last One Like This
@1
ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@2
GTGGCGTGAGTGCGGATCGATCGATGATCGATTCGATCGATCGCGCGAAAATAGAGCTAGCCC
@3
TCTTGATCGATCGATCGCTATTAGACGGCATGATCGATTCGATCGATCGCGCGAAAATATAGC
@4
ATCGTACCAGTACATACGATAGCCTCGTGATTCGATCGATGTGGCGTGAGTGCGGATCGATCG
@5
GTGGCAATATAGTGAGAGTAGAGTGCGGATCGAAATCGCTATCTAAGCTCAGTCGATTCTCTA
@6
CTAGACAGCTACAGTCCATGCGTGGCGTGAGCGATAGCTATCGATGACTCGATTGCGGATCGG
@7
GGTGAGTGCGGATCCGCTAGCTACCACATACGGGATAAAGCTAGTCTCTACGACGGTAGAGTA
@8
AGCTACGATTTATATCGGCTAGCCGTTATATAGCCGGACACAGATAGTACACAACAGTAGAGT
@9
GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC
@10
GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
When clean_data function is run, the label is always ? if no label is met. On first iteration the label is set to @1
which is fine, on second iteration, we have our nucleotide characters so we set them as values for our @1 label hence
'@1':'ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA' but here is a catch, on the third iteration,
we have a newline \n plus nothing '' so this block is run else: hence setting the '@1' to the stip()ed
value (technically now is nothing \n = '') which leads to override hence this key:'' on all iterations except
the last part @10which the code will act normally since there are no \n plus '' after the nucleotide characters.
4. Imagine Another Second Last One Like This
@1
ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@2
GTGGCGTGAGTGCGGATCGATCGATGATCGATTCGATCGATCGCGCGAAAATAGAGCTAGCCC
@3
TCTTGATCGATCGATCGCTATTAGACGGCATGATCGATTCGATCGATCGCGCGAAAATATAGC
@4
ATCGTACCAGTACATACGATAGCCTCGTGATTCGATCGATGTGGCGTGAGTGCGGATCGATCG
@5
GTGGCAATATAGTGAGAGTAGAGTGCGGATCGAAATCGCTATCTAAGCTCAGTCGATTCTCTA
@6
CTAGACAGCTACAGTCCATGCGTGGCGTGAGCGATAGCTATCGATGACTCGATTGCGGATCGG
@7
GGTGAGTGCGGATCCGCTAGCTACCACATACGGGATAAAGCTAGTCTCTACGACGGTAGAGTA
@8
AGCTACGATTTATATCGGCTAGCCGTTATATAGCCGGACACAGATAGTACACAACAGTAGAGT
@9
GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC
@10
GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
When clean_data is run, you may expect that the situation above may happen but remember, on first iteration the label
is @1 and the values are set to be '' (nothing) which maybe painful, but it has no pain this time since on next
iteration, we have our nucleotide characters that will override the '' (nothing) hence
'@1':'ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA' and this goes on till the last iteration.
5. Imagine Another Last One Like This
@1
GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
@2
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUACGUACGUAGCUGCGCGUAUUAUUACGAGAUAGACAGAUAGACAGAUCCUGACGAUCACG
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUAUUUAAUACAGAUCCUGACGAUUAAUACAGA
@3
AAAUGGAUAGUGAUGCUACGCUACGUAGCAUACGUUAUAUAUUACGUCGUAUGCUGACGUAUGAGAUAGACAGAUAGACAGAUCCUGACGAUCGUU
ACUACAGAUACCGAUCGAUAUAUCUGAUCCUGAUAGCAUUAUCUGAUCCUGAUAGCAUUAUUUAAUU
def clean_data(filepath, label_char):
"""
Receives a file path and separates strand labels from DNA
Returns a dict of {strand labels: DNA}
"""
DNAdict = dict()
# Temporary Storage For Read Indexes
label = '?'
try:
with open(filepath, 'r') as file:
# Set 'File Pointer' To Start At Zero
file.seek(0)
for i in file.readlines():
i = i.strip()
if label_char in str(i):
label = i
DNAdict[label] = ''
else:
DNAdict[label] = ''
DNAdict[label] += i
return DNAdict
# Any Exception Is An Error
except Exception:
return None
New updates so far like setting the 'File Pointer' to zero (start) of file. When this clean_data function is run,
it reads in the file and starts the for loop looking for the label_char. In a scenario like ours above, it will find
the label_char on first iteration as @1 then create a key for it having its value as '' hence '@1':''. On second
iteration, it finds the nucleotide characters and then enters the else block so it sets again our '@1':'' to '@1':''
and sets(hear me loudly on this point, it sets not adds) the current nucleotide characters. On third iteration, it
finds more nucleotide characters, it first checks is our @1 in these nucleotide and no so it goes to our else
block which sets our '@1':'GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
' to @1:'' and then adds the nucleotide characters to it hence '@1':'UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
'. In simple terms this line DNAdict[label] = '' in the else block makes our code ‘forget’ our last nucleotide
characters hence remembering the last bit of the nucleotide characters.
Hopefully you can figure out the bug now and how to navigate it before I can show you how I did it.
GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
@2
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUACGUACGUAGCUGCGCGUAUUAUUACGAGAUAGACAGAUAGACAGAUCCUGACGAUCACG
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUAUUUAAUACAGAUCCUGACGAUUAAUACAGA
@3
AAAUGGAUAGUGAUGCUACGCUACGUAGCAUACGUUAUAUAUUACGUCGUAUGCUGACGUAUGAGAUAGACAGAUAGACAGAUCCUGACGAUCGUU
ACUACAGAUACCGAUCGAUAUAUCUGAUCCUGAUAGCAUUAUCUGAUCCUGAUAGCAUUAUUUAAUU
On such a file format, our function still works the same as above (remembering the last bit of the nucleotide characters)
but the first iteration is where I will shine light upon. So on first iteration, we do not have a label and so by default,
this ? the label if label is not overridden. Our function enters if statement to check if our i (now nucleotide
characters) has our label_char hence no then else block is into play and for the first time makes ? as '?':''
and adds nucleotide characters to it hence '?':'GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
' and on the next iteration it will forget the previous by setting again hence our '?':'GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
' becoming '?':'UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
'. This goes on and on till last iteration.
@1
GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
@2
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUACGUACGUAGCUGCGCGUAUUAUUACGAGAUAGACAGAUAGACAGAUCCUGACGAUCACG
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUAUUUAAUACAGAUCCUGACGAUUAAUACAGA
@3
AAAUGGAUAGUGAUGCUACGCUACGUAGCAUACGUUAUAUAUUACGUCGUAUGCUGACGUAUGAGAUAGACAGAUAGACAGAUCCUGACGAUCGUU
ACUACAGAUACCGAUCGAUAUAUCUGAUCCUGAUAGCAUUAUCUGAUCCUGAUAGCAUUAUUUAAUU
@4
When Our function is run on this one, it runs normally but on the last part is where I will shine light upon. The second
last iteration, we have our label as @4 and there are no nucleotide chars but there is a \n and ''. Our function
uses Python’s strip() function that removes \n\t\r and so much more hence we are left with '' and this is set to
@4 hence '@4':''.
Here is the revised code
def clean_data(filepath, label_char):
"""
Receives a file path and separates strand labels from DNA
Returns a dict of {strand labels: DNA}
"""
DNAdict = dict()
# Temporary Storage For Read Indexes
label = '?'
try:
with open(filepath, 'r') as file:
# Set 'File Pointer' To Start At Zero
file.seek(0)
for i in file.readlines():
i = i.strip()
if label_char in str(i):
label = i
DNAdict[label] = ''
else:
if label not in DNAdict.keys():
DNAdict[label] = ''
DNAdict[label] += i
return DNAdict
# Any Exception Is An Error
except Exception:
return None
In the else block, I added an if statement to check if the label is not in the dict() keys, if not then create it
one key:value (the key being ‘?’ and value is whatever nucleotide characters we find) else jump the if statement. Also
added the try and except blocks so that any exception is an error.
Note: Let me know if you have any better versions for this function.
user_home.py
The variables, functions or modules without declarations are handled in the class that is in the file
def start_DNA_analysis(self, filename):
if validate_file(filename) == 0:
DNA_label_entry = CTkEntry(self.user_home_window, placeholder_text="Enter DNA label",
font=("Menlo", 20), fg_color="black", text_color="green",
height=30, width=300, border_width=5, border_color="orange",
placeholder_text_color='cyan')
DNA_label_entry.place(rely=0.915, relx=0.09)
seq_type_entry = CTkEntry(self.user_home_window, placeholder_text="Enter DNA or RNA",
font=("Menlo", 20), fg_color="black", text_color="green",
height=30, width=300, border_width=5, border_color="orange",
placeholder_text_color='cyan')
seq_type_entry.place(rely=0.915, relx=0.45)
submit_DNA_label_button = CTkButton(self.user_home_window, text="Submit", font=("Menlo", 25),
text_color="blue", corner_radius=5, fg_color="black",
command=lambda: DNA_analysis(filename, str(DNA_label_entry.get()),
str(seq_type_entry.get()).upper(), self.user_home_window, self.username,
self.firstname, self.lastname, User_Home))
submit_DNA_label_button.place(rely=0.915, relx=0.8)
return
else:
warning_2 = CTkLabel(self.user_home_window, text="File Problem", font=("Menlo", 30), fg_color="black",
text_color="red")
warning_2.place(rely=0.87, relx=0.4)
self.user_home_window.after(1000, warning_2.destroy)
return
When you use the proper version, even some of the file formats that are forbidden (as shown above). This part of code will run in order to get the read index (the text tagged on each sequence read) from the user.
1. Imagine Something Like This
After selecting a file to read and analyze, the widget for getting the read index (the text tagged on each sequence read)
pops up and asks the user for the read index (the text tagged on each sequence read). The user well instead of putting in
the most-common character in the read indexes (like for our scenario files, the most is @), the user puts in @1 or
chooses @ plus some of the other numbers (like @2 and so on). Remember our function in DataTools.py that cleans
the file data.
That clean_data function will get the label we put (for instance @1). The function will look for only this (@1) and
add stuff (this time both the read indexes and the nucleotide characters because they are not the label) hence getting key
read index (@1):nucleotide characters then read index:next other read index at respective intervals till read index
@9 where it all happens, here at the nucleotide characters it gets @1 and @9’s nucleotide chars hence
'@1':'GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC'.
You may ask why it happened, well, remember that we store our data as strings (flexible, easy error checking and convenient)
and next read index has @1 in it and the read index the user put was to find anything with @1 in it and @10 has @1
in it hence @1:'GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC' and then
@10:GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT because the code made a new key (because it had @1
in it) and gave it the next nucleotide characters GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
2. Imagine The Next Scenario Like This
A user puts in @2 as the most-common read index. The first iteration, the loop finds @1, but @1 doesn’t have @2
in it so the code is makes it a ? and then will add the next line of nucleotide characters. On second iteration, the loop
finds @2 and it sets @2 as key and adds the next line of nucleotide characters. As the iterations go on and overwrites
happen it will reach the last line of nucleotide characters hence adding that and ending the loop.
hamming.py
1. Resources, If Statements and Logic
Imagine I have a good valid file format like this
@1
GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
@2
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUACGUACGUAGCUGCGCGUAUUAUUACGAGAUAGACAGAUAGACAGAUCCUGACGAUCACG
@3
AAAUGGAUAGUGAUGCUACGCUACGUAGCAUACGUUAUAUAUUACGUCGUAUGCUGACGUAUGAGAUAGACAGAUAGACAGAUCCUGACGAUCGUU
Then a snippet of code in the massive code base like this
def show_hamming(self, DNA_data):
""" Showing Hamming Distance """
temp_seq_1 = str()
temp_seq_2 = str()
# Getting Sequences From Data
for key, value in DNA_data.items():
if key == str(self.seq_1_entry.get()):
temp_seq_1 += str(value)
elif key == (self.seq_2_entry.get()):
temp_seq_2 += str(value)
A briefing on If Statements, If statements are clauses in code that check conditions, whether True or False. In a
scenario above, the if statement starts in the loop and let’s say maybe user entered @1 as read index 1 and @1 as
read index 2 for Hamming Distance (Hamming distance checks index by index for not matching nucleotides while respecting
the index, if I am checking index 1 of seq 1 then I must check index 1 of seq 2 too), it checks (is key == @1 and yes)
so it sets temp_seq_1 to the nucleotide characters for @1 and the if statement breaks out (like it doesn’t check again)
so we leave the iteration with @1 and go to the next iterations that have other keys (not @1) hence if seq 2 is @1.
The shot of @2 is lost.

But on code like this
def show_hamming(self, DNA_data):
""" Showing Hamming Distance """
temp_seq_1 = str()
temp_seq_2 = str()
# Getting Sequences From Data
for key, value in DNA_data.items():
if key == str(self.seq_1_entry.get()):
temp_seq_1 += str(value)
if key == (self.seq_2_entry.get()):
temp_seq_2 += str(value)
The two If statements run because all are ‘primary’ (they must run since they start the ‘check conditions system’) hence no errors.
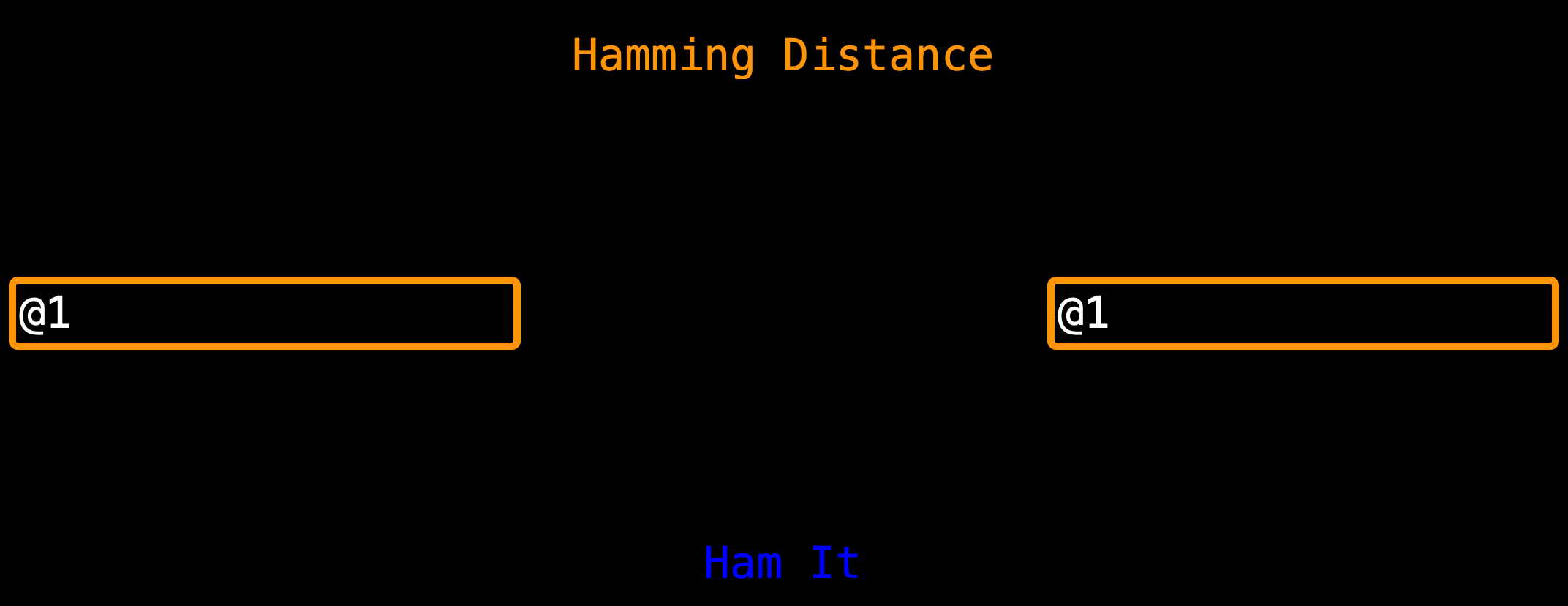
Note: If You really love scenario 1 and it is useful to you (scenario 1 uses less computer resources) while scenario 2 will take more time on big data and secondly, it is known that a sequence compared to itself is a photocopy and I love optimization hence I left scenario 1 as default. Let me know which is best.
Usage
Big Note
In computers, counting starts at 0 (zero) but to simplify this for you I have added 1 (one)
to the GUI Display but left it as 0 (zero) in the computer processes so that you can see
that as usual we count from 1 (one) yet relative to the computer, it started at 0 (zero) but
relative to you, it starts at 1 (one).
Hence; If you want the first position of anything, just use 1 (one) in the GUI
So use the numeracy on the screen and do not assume!
1. File Format
@1
ATGGCGTGAGTGCGGATCGATCGATGATCGATCGATCGCTATTAGACGGCATCGATCGATCGA
@2
GTGGCGTGAGTGCGGATCGATCGATGATCGATTCGATCGATCGCGCGAAAATAGAGCTAGCCC
@3
TCTTGATCGATCGATCGCTATTAGACGGCATGATCGATTCGATCGATCGCGCGAAAATATAGC
@4
ATCGTACCAGTACATACGATAGCCTCGTGATTCGATCGATGTGGCGTGAGTGCGGATCGATCG
@5
GTGGCAATATAGTGAGAGTAGAGTGCGGATCGAAATCGCTATCTAAGCTCAGTCGATTCTCTA
@6
CTAGACAGCTACAGTCCATGCGTGGCGTGAGCGATAGCTATCGATGACTCGATTGCGGATCGG
@7
GGTGAGTGCGGATCCGCTAGCTACCACATACGGGATAAAGCTAGTCTCTACGACGGTAGAGTA
@8
AGCTACGATTTATATCGGCTAGCCGTTATATAGCCGGACACAGATAGTACACAACAGTAGAGT
@9
GCTCGATAGAGTCCAGATCCATCAGACAGGGAATATATTACAGATACAGGGAGGTAGAGAAAC
@10
GTTACACAAGGATCGCTACAGATATCGGTACGCTAAATATCGCGCCTTAGTAGAGTCGAGTGT
OR
@1
GACUACAGAUACCGAUCGAUAUACGACAGAUACAGAUAGACAGAUAGACAGAUCCUGACGAUCAGAUAGACAGAUAGACAGAUCCUGACGAUCUUA
UCAGCGAGCAUCUGAUCCUGAUAGCAUGCUGACGUAUGAGAAUUUAAUCGGUACGUACGUAGCUGCG
@2
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUACGUACGUAGCUGCGCGUAUUAUUACGAGAUAGACAGAUAGACAGAUCCUGACGAUCACG
UCAGCGAGCAUCUGAUCCUGAUAGCAUUUAAUCGGUAUUUAAUACAGAUCCUGACGAUUAAUACAGA
@3
AAAUGGAUAGUGAUGCUACGCUACGUAGCAUACGUUAUAUAUUACGUCGUAUGCUGACGUAUGAGAUAGACAGAUAGACAGAUCCUGACGAUCGUU
ACUACAGAUACCGAUCGAUAUAUCUGAUCCUGAUAGCAUUAUCUGAUCCUGAUAGCAUUAUUUAAUU
File Formats are key for good results, so I recommend a file with this format fastq,fasta or a txt file with this
format for nice and clean results and not to encounter some of the bugs that are in the Bugs Found section.
2. Sign Up Tutorial
 When signing up, avoid commas because I used them for separating data in the database. What happens in most cases is that
when they are passed, they hinder the healthy retrieval of data from the database hence causing injections, data breaches
and so much database problems. I have put good measures to prevent this by not allowing commas from users.
When signing up, avoid commas because I used them for separating data in the database. What happens in most cases is that
when they are passed, they hinder the healthy retrieval of data from the database hence causing injections, data breaches
and so much database problems. I have put good measures to prevent this by not allowing commas from users.
Note: If you are a security expert or have a better way, please reach out to me via the contacts in the contacts section.
3. File Creation and File Reading tutorial
 When entering a folder name to create, by default I have redirected the folder creation to the
When entering a folder name to create, by default I have redirected the folder creation to the Desktop. All created
folder names without a full path (absolute file path) to where to create them will be put in the Desktop. If there is an
absolute file path, that is where the folder creation will happen.
When reading a folder, it is encouraged to enter the absolute folder path to where the folder is hence reading it healthily. If you do not provide an absolute folder path, the program with error at you.
4. Is It A DNA File, RNA File OR Not
 I have simple function that reads in a file pressed and counts the
I have simple function that reads in a file pressed and counts the A, T, G, T, U. If those letters are greater than the
other stuff in the file, then the file would be valid to be analyzed.
Note: But file validation is not yet over.
5. Read Index Usage
 When entering the
When entering the Read Index, please enter a character that is most-common in all the read indexes for better and
reasonable results.
This where part two of file validation occurs, when you enter the file, it will also ask you to enter a sequence type
either it is DNA or RNA. If all do not match what it has from the file then you cannot proceed to analysis.
6. Codon Usage Entry
 When entering a Read Index to get the codon activity, just enter a number (like
When entering a Read Index to get the codon activity, just enter a number (like 1 or 2) this is used as a reference
that the user wants codons in read index number 1 or read index number 2. Do not use @1 or @2 depending on what
read index you want, just use 1 or 2 or whatever read index you want but without other characters (in this case those
characters are @ just type in a number)
Note:
1+ means that is a normal read of that sequence
1- means that is a reversed complement read of that sequence
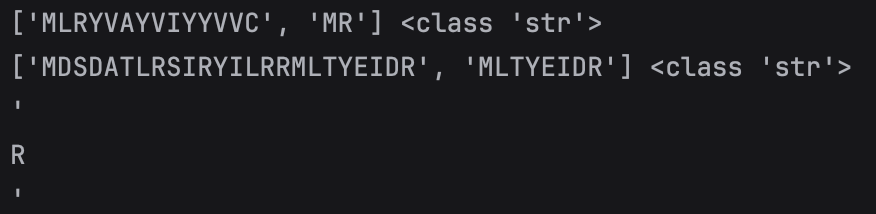
7. Protein Interpretation
Display of Proteins would look messy if I displayed proteins by finding the instance of M (start Codon) without seeing
that there are proteins inside of proteins. For instance as in the picture, a long protein is shown but not till its _
(stop Codon). Since the code has this if letter_6 == "M":, it will cut proteins short hence not seeing the full picture.
Since I knew this was a string (it is NOT a list), I added this line of code
if letter_6 == "M" and proteins[i_6 - 1] == '\'':. The long protein has a protein subunit but with the new code inserted,
the long protein is shown and the next “protein(s)” is a protein subunit(s) of the long protein above it.
Hence

Note: Once again
1+ means that is a normal read of that sequence
1- means that is a reversed complement read of that sequence
Note: My proteinsynthesis function does not show partial proteins (proteins that have a start but have no stop codon downstream) but only shows full proteins (proteins that have a start codon and a stop codon). Which means that I used open reading frames NOT reading frames.
8. Proteinsynthesis Time Calculation

My timer can be off by some amount of time because of factors like laptop updating time late due to it finishing other tasks and mostly due to using python threads which can be interrupted by python’s Global Interpreter Lock (GIL). The GIL allows one thread to execute at a time which boosts the interpreter’s speed but giving up true parallelism of processes.
Note: For just keeping track of time and proteinsynthesis, I decided to use threads (easy to understand).
9. Hamming Distance
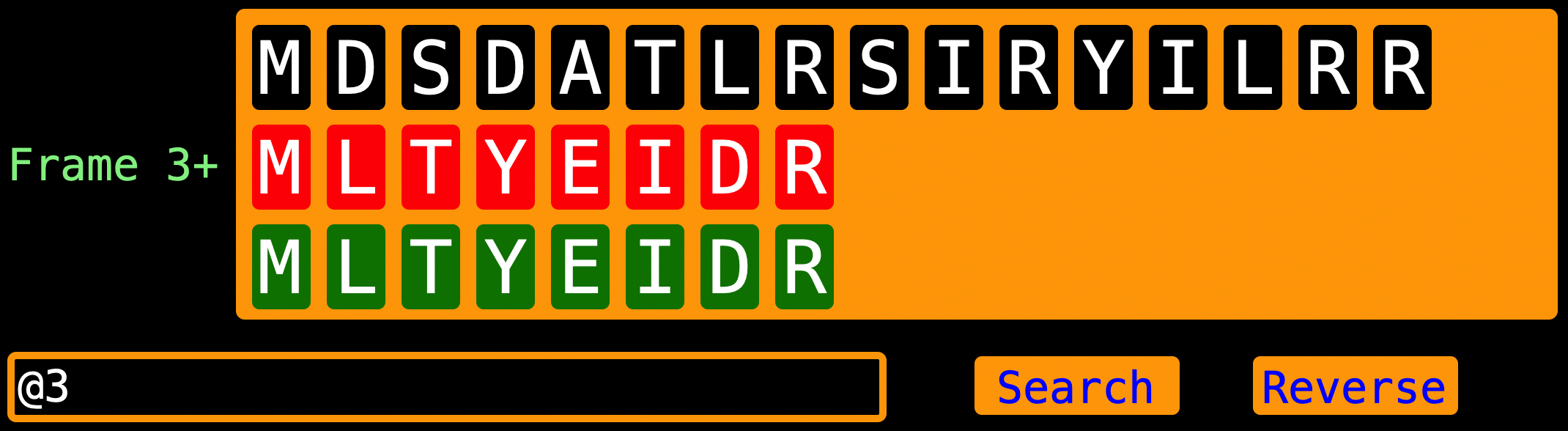
 When entering the read index, enter the actual read index (for our scenario, the read index would be
When entering the read index, enter the actual read index (for our scenario, the read index would be @1) depending on
which indexes you want to “ham”.
Note: LengthOfEach - is the length of the current view and its analysis | is the entire
length of each of the strands
10. K-mer By Search
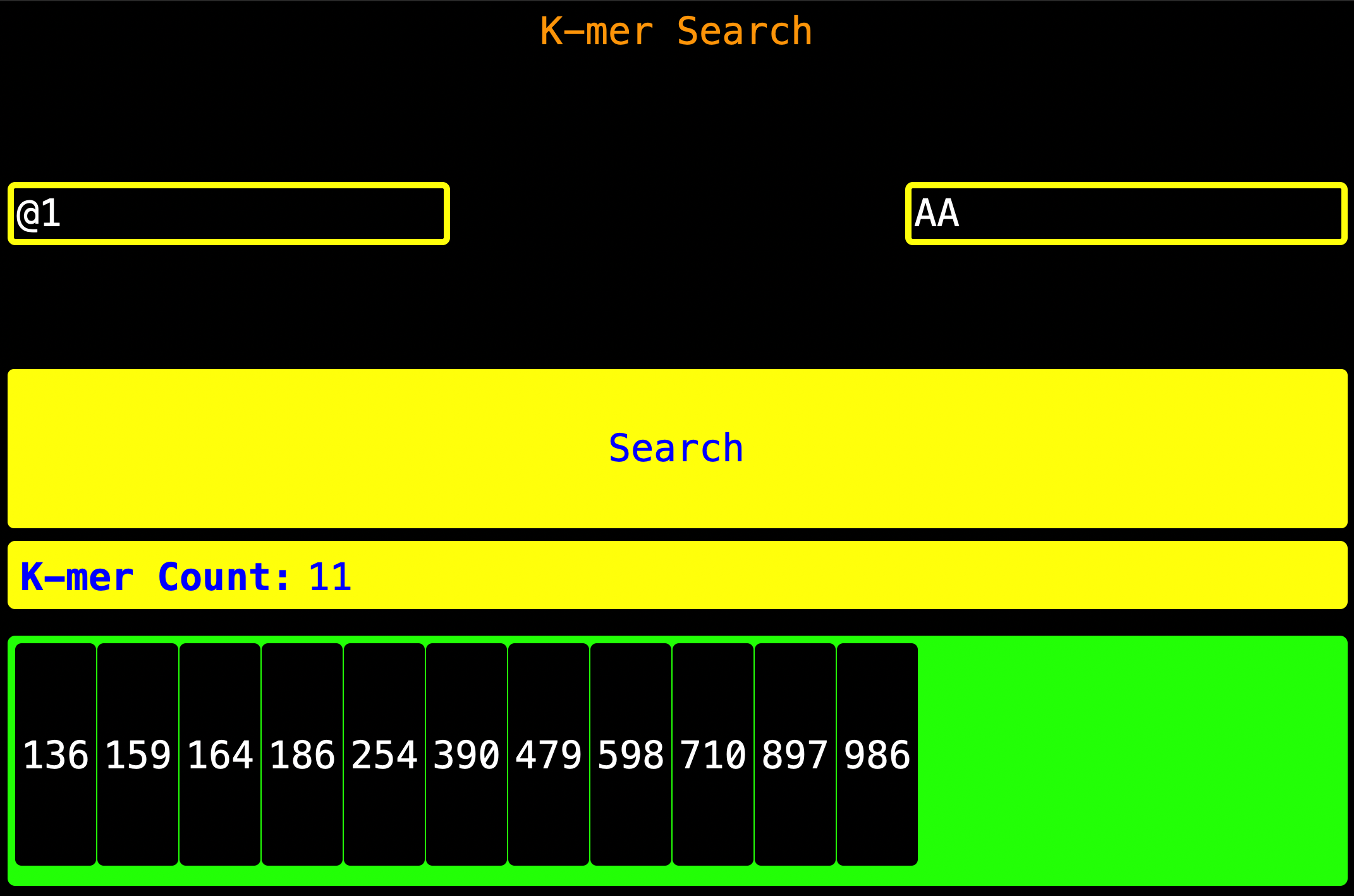 As you can see, enter the
As you can see, enter the Read Index as shown on the left of the screen, enter the K-mer to be found as shown on the right
then press Search. Once search is pressed, the k-mer Count will be updated and the indexes where the K-mer is found will
be found will be shown.
K-mer Count - This is the number of times the k-mer appeared
App Hack:
You can see how many nucleotides are there in the `Read Index` by entering the
nucleotide base (like `A` in order to get how many `A` are there)
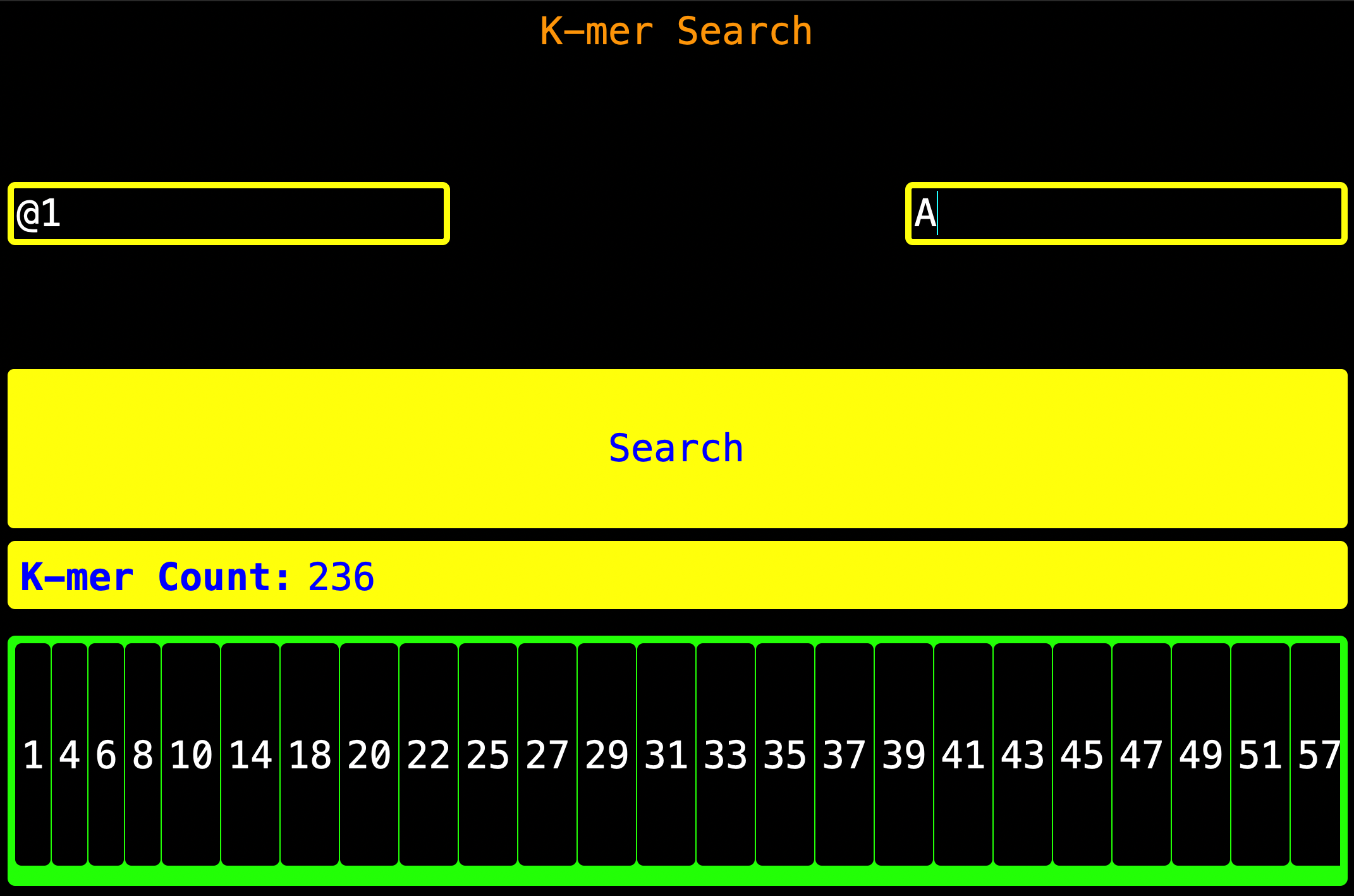
Note: The numbers shown in the visualization panel are the index where the K-mer starts then add the K-mer’s length
(the K-mer’s position inclusive - if the K-mer length is 5, then just add 5 to the index and **subtract 1 hence you
get the stop position) forward and that is where the K-mer stops.**
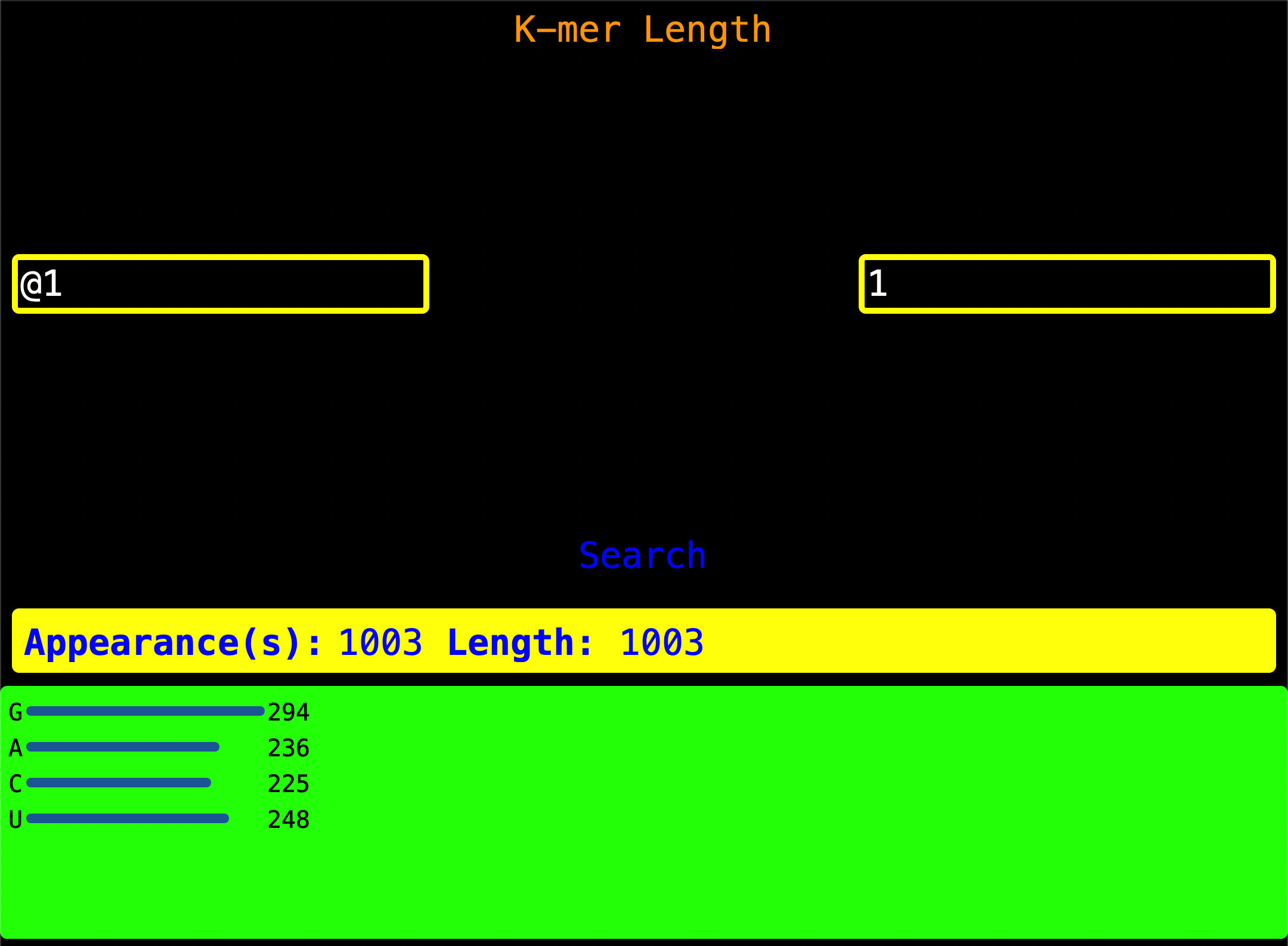
11. K-mer By Length
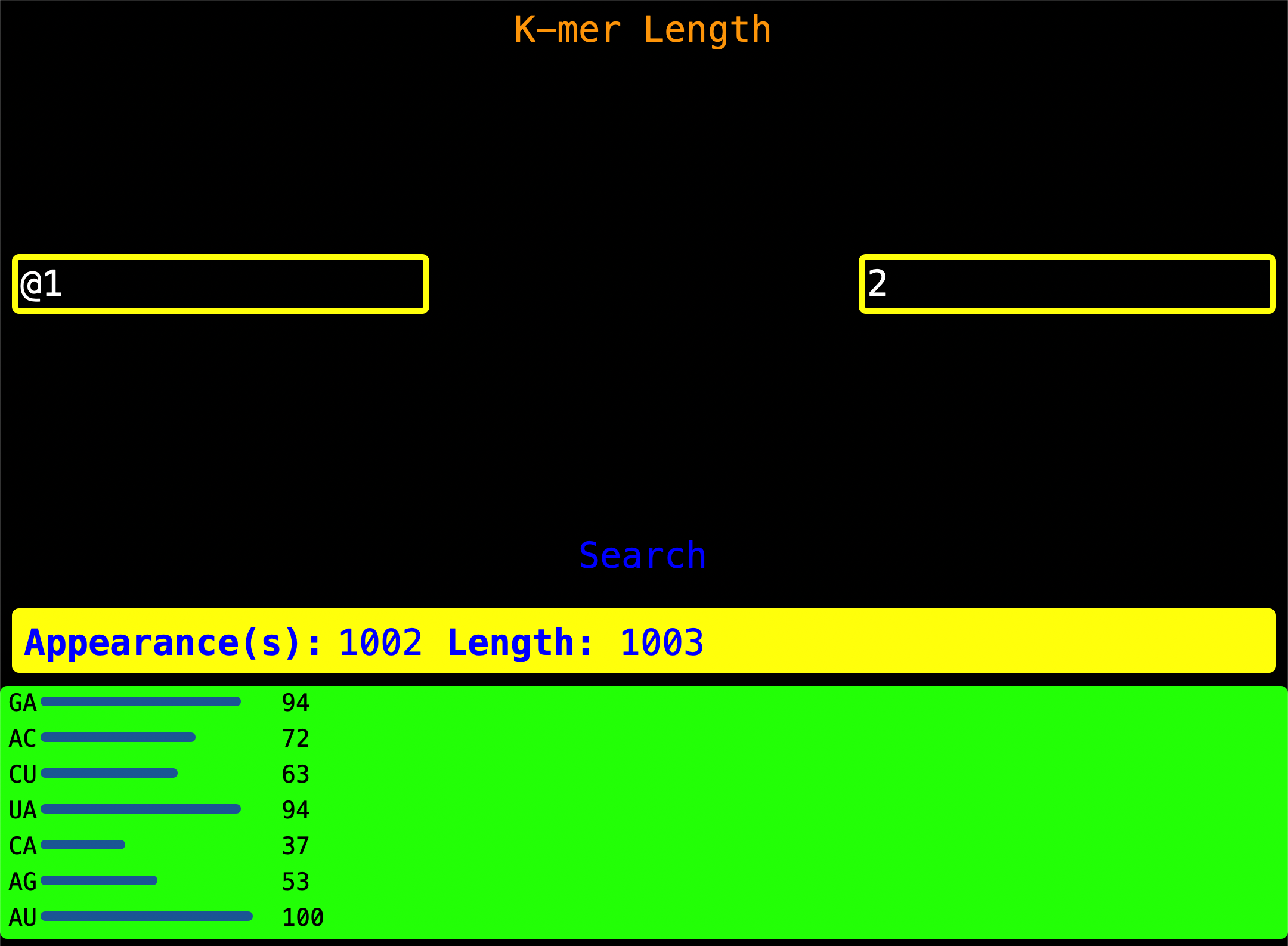 Enter the Read Index as shown on the left of the screen and enter a valid k-mer length on the right of the screen. When
Enter the Read Index as shown on the left of the screen and enter a valid k-mer length on the right of the screen. When
Search is pressed, it looks for all two-longed k-mer and gives you a visualization of k-mer by length.
Appearance(s) - This is the number of times a two-longed k-mer or n-longed k_mer was found.
Length - Length of the sequence.
App Hack:
You can see the nucleotide count for a Read Index by just asking the program for all
single-longed nucleotides characters hence getting all nucleotides and their count
Decode
Decode is a primitive programming language that will help users complete more custom tasks that are not premade in the
app. In this case it has just features like setting variables comments mathematical operations like
addition subtraction division multiplication and loops logcial statements printr. All of these can help a user
get a simple task done like GC Content Nucleotide Count AT Content Some Mathematical Algorithms.
Cannot Handle Decimals
Let’s say user starts by setting 5.4 to a variable called x then sets 0.0 to a variable y that will later store our result. When user tries to divide the value in x by 3.2, Decode will error saying that 3.2 is not an integer.
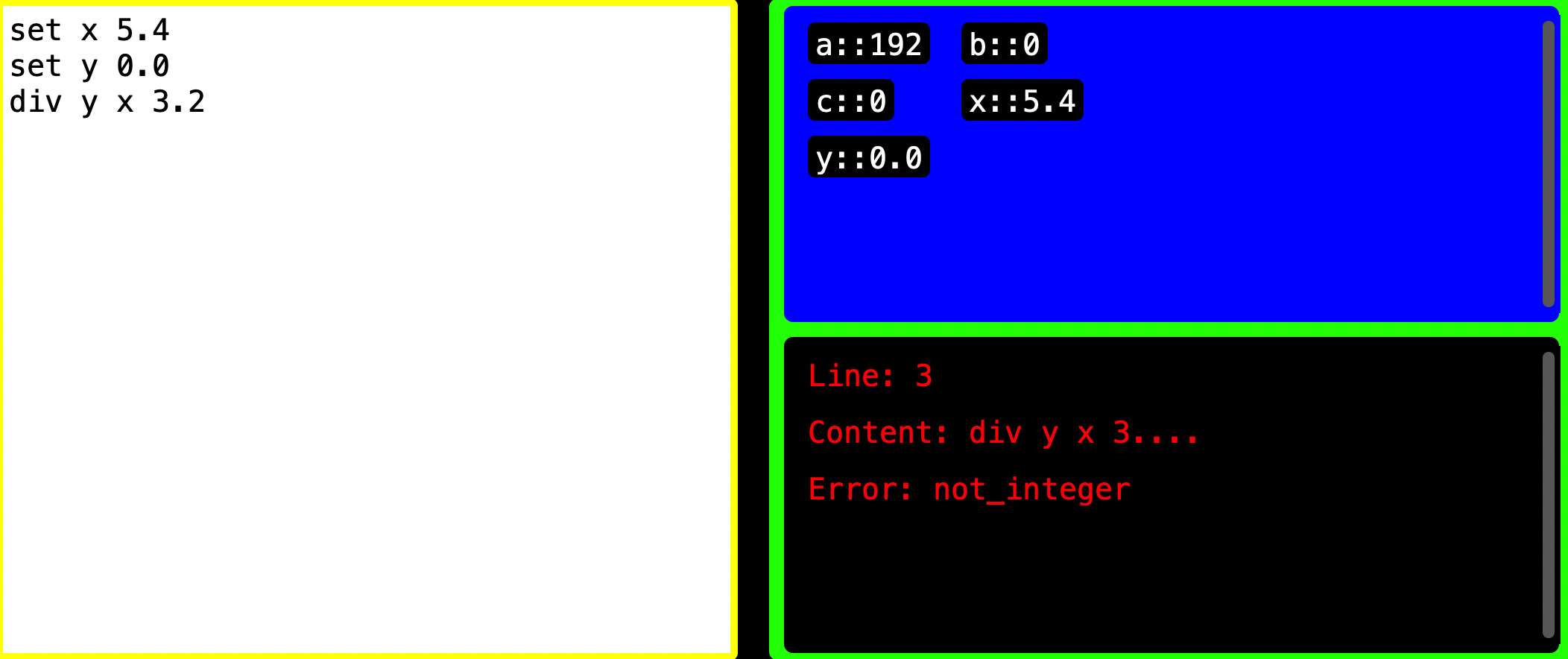
REASON: Every variable receives a string version of the value user gives it. When you set 5.4 to x, Decode thinks it is
string version of 5.4 which is 5.4 into x hence no error. When user divides x by 3.2, Decode recognizes that these are not
integers and errors saying not_integer
Let’s say user sets a variable called x which we will be our result variable and set it to 0. Later user tries to divide
3 by 2 which will result into a decimal. Decode uses integer division in division (quotients are stripped of their
decimal part and made integers by leaving the floor - the whole number like 3 divided by 2 is 1.5 but the .5 is removed
and the 1 is left as the answer)

REASON: Decode first checks if the divisor and the dividend are integers and proceeds and uses integer division
Cannot Handle Nested Loops
Let’s say user sets two loops, one outside and one inside the other forming nested loops.

When when is called, its program counter or location is stored and used as a reference to start from next time the loop
needs to start again if condition is not met. In this scenario the first when location is saved and the loop goes on but
another when is encountered which overwrites the previous when saved location. Since this current when is the one
remembered, its loop in run and when finished, the program breaks out and continues. When the outer loop’s end is found,
the other loop that ran overwrote or reset important variables needed for this end to execute hence this end is skipped
and program continues without errors. The program does not run as intended since one loop can run at a time NOT loop
inside a loop.
Cannot Handle Nested If Statements Logically
If statements have many twists and turns. In this scenario user sets up an if statement inside an if statement. An if
statement does not need to save anything so no worry at the if k 1 part but when Decode reaches done it checks if the
program is running, if it is then done is skipped. If the program is NOT running, done will release the program from
not running to running and if there is code following then that code will always be run.

Infinite Loops
In when d Decode is checking if d is zero and if not, then the program will run forever or indefinitely. The app will
stop responding and the program will go on. Threading is a useful library that allows running more than one process at the
same time. I made the RUN button start the “child” thread that runs the code while the “main” thread runs the GUI interface.
I added a STOP button that ends the “child” thread hence the GUI interface runs normally and the code is run normally at
the same time.

Example Usage For Decode Code That Shows Only “AT” Nucleotides In DNAGenre
set x a
// AT counter
set y 0
when x
if - A
branch 15
done
sub x x 1
add c c 1
end
if c a
stop
done
printr 1
add c c 1
if c a
stop
done
if - T
printr 1
sub c c 1
add y y 1
branch 9
done
sub c c 1
printr 0
branch 9
y counts how many times AT has been found in the sequence but never considers the last A because that last A
is NOT followed by a T
In a scenario like AA, TT, GG, CC or other similar k-mers, the algorithm cannot highlight the k-mers logically.
Note: As I was doing these 33 lines of code, I realized after debugging that; checking bounds is key; my language is verbose; and spacing makes the language more readable. It was tough to write this one. I know it is worse than Assembly but at least it got a simple task done.
Tech Stack
Pycharm - Code Editor
ChatGPT - AI Assistant
I am very fluent with prompting most Large Language Models(LLMs) because I use patterns in my prompts, simple prompts and most notably use words that cause attention hence push AI to reason.
For example; I want ChatGPT to give me an essay for a college application
I would say
Hey, What's up, I would love a well-polished college essay like those college essays in
the early 2000s.
Instructions
1. Use simple English
2. Use less words
3. Use 2025 college essay structure
Explanation;
When I say Hey, What's up the model tunes its attention to a ‘sweet mode’ and will reason nicely and happily (due to
inbuilt ethical restrictions you may see that when it is nice but not when it is angry)
When I add I would love a well-polished college essay the word I would love sharpens it niceness and the word
a well-polished essay makes it pick a good essay out of those in it brain.
When I add like those college essays in the early 2000s it takes the models attention to college essays written in
the early 2000s (I do this because college essays by then have changed over time and this makes humans not even close
to thinking it was AI). It will use the slang and language by then hence I can edit the output essay to an appropriate level.
Use simple English this amplifies the 2000 timeline produced college essay
Use less words this makes the model use less word but if I said Use 200-500 words, I am using the data of today mostly
which will bring it back to 2025, yet I want something way back like 2000s.
Use 2025 college essay structure this will bring it back to 2025 after carrying everything from 2000s (our college essay)
and just make it look like 2025.
Here is what ChatGPT gave me, no reasoning just ChatGPT (great for everyday tasks)
When I was twelve, I broke apart my family’s old computer. Everyone thought I had ruined it,
but I wasn’t trying to destroy anything. I wanted to know how it worked. The wires, the
circuits, the dust inside—it all looked like a puzzle waiting to be solved. I didn’t fix
the computer that day, but I fixed something in myself: my fear of breaking things.
Since then, I’ve treated learning as a kind of tinkering. When I study math, I pull apart formulas
to see how each step fits. When I write code, I test lines until they fail, then rebuild.
Even in conversations, I ask questions that make people pause, as if I’ve unscrewed a piece
of their thought. For me, mistakes are not dead ends—they are the screws on the floor,
proof that I’m working toward understanding.
Still, curiosity can be lonely. Friends sometimes tease me for “thinking too much.”
But I’ve learned to see this as a strength. It’s what helps me stay with a hard problem until it finally clicks.
It’s also what makes me want to study computer science in college: a field where asking “what if” is not only allowed,
but required.
I want to be the kind of person who doesn’t just use technology but shapes
it—who builds tools that make life simpler, smarter, or fairer. I know I’ll keep breaking
things apart, whether it’s code, ideas, or assumptions. But now, I break them with
purpose: to build something better.`
Contributing
If you are interested in improving or upgrading this project even though you are a beginner or expert,
please feel free to contact me through my contacts in the contacts section.
Copyright
If anyone wants to use this project for their project(s), education or work, please do freely.
Please feel at ease to either star the project or fork it just to show an appreciation
(It is optional to star or fork).
Contact
It is very convenient to reach out to me through these contacts below because I can answer
within minutes.
LinkedIn - My LinkedIn